Chemioterapici Antineoplastici
Attualmente
disponiamo di svariati farmaci che possono essere usati con vantaggio nei vari
tipi di malattie, circa un centinaio, che vengono comunemente chiamate cancro.
Sono raggruppati in sette classi
·
Antimetaboliti
·
Farmaci ad
elevata reattività chimica
·
Farmaci
complessanti il DNA
·
Antimitotici
·
Ormoni
·
Enzimi
·
Farmaci vari
a
loro volta suddivise per gruppi (Tabella 1)
Tabella 1
|
1. Antimetaboliti |
1.1 Antagonisti dell’acido folico: Mtx,
Aminopterina 1.2 Antagonisti pirimidinici: FU
e analoghi, Ara-C 1.3 Antagonisti purinici: MP,
TG, Azatioprina 1.4 Inibitori della
ribonucleosidodifosfatoreduttasi: Idrossiurea 1.5 Antagonisti della glutamina: Azaserina, DON |
|
2. Farmaci provvisti di elevata
reattività chimica ma privi di specificità di azione |
2.1 Mostarde azotate: Mecloretamina,
Melfalan, Clorambucile, Ciclofosfamide, Trofosfamide 2.2 Aziridine: Tiotepa,
Mitomicina C 2.3 Altri alchilanti: Busulfan,
Pipobroman, CCNU, Dacarbazina, |
|
3. Farmaci che complessano il DNA |
3.1 Actinomicine: Actinomicina D 3.2 Bleomicine: Bleomicina A2,
Bleomicina B2 3.3 Antracicline: Daunomicina,
Adriamicina |
|
4. Inibitori della mitosi |
4.1 Alcaloidi della Vinca: Vinblastina,
Vincristina 4.2 Taxani: Paclitaxel,
Docetaxel ed altri agenti che stabilizzano i microtubuli |
|
5. Ormoni |
5.1 Steroidi: Testosterone, Calusterone,
Idrossiprogesterone, Medrossiprogesterone, Ciproterone 5.2 Ormonoidi: Dietilstilbestrolo,
Tamossifene |
|
6. Enzimi |
6.1 L-Asparaginasi |
|
7. Farmaci vari |
7.1 Cis-platino, Irinotecan,
Topotecan |
1. Antimetaboliti
La sintesi proteica è il risultato della trasmissione dell’informazione contenuta nel DNA ad un RNA, che la trasferisce quindi alla proteina. Questo è, secondo una definizione coniata da Crick, il dogma centrale della biologia.
La sintesi del DNA richiede la contemporanea presenza di quattro desossinucleotidi trifosfato (desossiadenosintrifosfato, dATP; desossiguanosintrifosfato, dGTP; desossicitidintrifosfato, dCTP; desossitimidintrifosfato, dTTP) e pertanto nella cellula che va incontro a mitosi vi deve essere una considerevole produzione di tali composti, che si formano per riduzione in 2’ del ribosio a 2’-desossiribosio.
Ora, poiché le cellule tumorali continuano a dividersi senza sosta e poiché la moltiplicazione cellulare richiede una sintesi netta di acidi nucleici, si è cercato di trovare composti che, inibendo selettivamente la formazione di acidi nucleici o di loro precursori, siano in grado di arrestare la crescita incontrollata della massa tumorale. Ciò è stato realizzato con gli antimetaboliti, farmaci capaci di interferire nel meccanismo di formazione o di utilizzazione di un normale metabolita cellulare, per esempio interferendo con le reazioni che portano alla formazione dell’anello pirimidinico (Tavola 1) o di quello purinico (Tavola 2), propri delle basi che costituiscono gli acidi nucleici.
Un antimetabolita può agire o inibendo uno o più
enzimi oppure sostituendosi al metabolita fisiologico a livello delle
macromolecole dell’organismo (acidi nucleici, proteine) fornendo prodotti
spuri, cioè non fisiologici.
Bastano piccoli cambiamenti nella struttura di un metabolita, per es. la sostituzione di un ossidrile con un gruppo aminico NH2 o l’introduzione di un gruppo metilico CH3, per trasformarlo in un composto con attività antagonista o antimetabolita. In realtà in tale cambiamento influiscono non solo la natura del sostituente ma anche le sue dimensioni e quindi la capacità o meno del nuovo composto di legarsi alla superficie recettoriale dell’enzima da inibire o di inserirsi come unità strutturale fraudolenta (cioè estranea) a livello degli acidi nucleici.
1.1 Antagonisti dell’acido folico
A differenza degli antagonisti della glutamina, che non hanno prodotto farmaci di uso terapeutico, nell’ambito degli antifolici abbiamo un farmaco di notevole interesse clinico: il metotressato (Mtx). E’ noto che l’acido tetraidrofolico sotto forma di cofattore (coenzimi F, acido folinico e congeneri) provvede al trasferimento dell’unità monocarboniosa. Tale trasferimento avviene due volte nel corso della biosintesi dei nucleotidi purinici (formazione dell’IMP) ed una volta nel caso della biosintesi dei nucleotidi pirimidinici (sintesi ad opera della timidilato sintetasi dell’UMP, che viene quindi trasformata in dTMP).
Esiste pertanto la possibilità di sintetizzare composti capaci di inibire la riduzione del diidrofolato a tetraidrofolato, necessario per la sintesi del cofattore folinico (CoF), colpendo così le cellule in mitosi, poiché solo in queste avviene la formazione della desossitimidina.
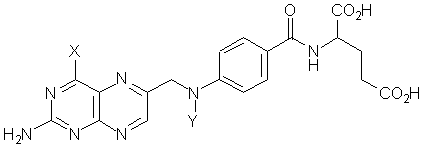
X Y
acido folico OH H
aminopterina NH2 H
metotressato NH2 CH3
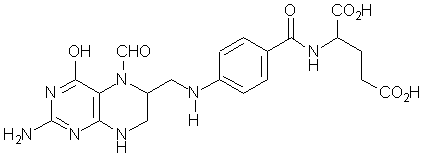
acido folinico (CoF)
Varie piccole modifiche sono state apportate alla molecola dell’acido folico per ottenere sostanze ad attività antifolica, ma dei vari derivati sintetizzati solo due hanno mostrato elevata attività anticancro: l’aminopterina e l’ametopterina, meglio nota come metotressato (Mtx). Ambedue questi antifolici presentano elevata tossicità, minore invero nel Mtx che ha soppiantato nell’uso clinico l’aminopterina, sia perché colpiscono tutte le cellule che si dividono rapidamente, come quelle della mucosa intestinale, sia perché impedendo in tutti i tessuti la formazione di acido tetraidrofolico a partire dall’acido diidrofolico (attraverso un legame quasi irreversibile con l’enzima diidrofolato reduttasi) riducono in essi il livello della forma coenzimatica (CoF). Il metotressato trova impiego nel coriocarcinoma femminile (70% di remissione) ed in modo particolare nella leucemia infantile acuta (35-50% di casi di remissione totale).
L’individuazione e lo sviluppo di nuovi analoghi più selettivi del Mtx è attualmente uno dei campi in cui più attiva è la ricerca farmaceutica relativamente agli antitumorali.
1.2 Antagonisti pirimidinici
Sul piano pratico si sono rivelate utili alcune fluoropirimidine, come il 5-fluorouracile (FU) ed il suo 2’-desossiribonucleoside o flossuridina (FUdR), che vengono ambedue metabolizzati ad acido 5-fluoro-2’-desossiribonucleico, un potente inibitore competitivo della timidilato sintetasi, l’enzima che normalmente converte l’uridinamonofosfato (UMP) in acido timidilico utile per la sintesi del DNA. Il 5-fluorouracile, sotto forma di FUTP può essere incorporato fraudolentemente nel RNA fornendo un F-RNA spurio ed incapace di funzionare. Gli enzimi che metabolizzano l’uracile (normale metabolita) prontamente degradano il 5-fluorouracile (antimetabolita) in prodotti di demolizione fluorurati, ma questo processo ha luogo solo nelle cellule normali, mentre le cellule cancerose non sono capaci di catabolizzare in questo modo il FU. Questo diverso comportamento potrebbe spiegare l’attività antineoplastica manifestata dal FU, che non potendo essere degradato esplica interamente la sua azione antimetabolica.
La flossuridina non presenta vantaggi dal punto di vista terapeutico ed il 5-fluorouracile è il solo derivato veramente utile in sede clinica, principalmente contro i tumori solidi del tratto gastrointestinale, della mammella e dell’apparato genitale femminile. Cosa piuttosto strana, non risulta attivo contro la leucemia ed i linfomi.
Recentemente ha destato interesse un analogo della flossuridina, il fluorofuran (Ftorafur, Tegafur). E’ al momento in fase di valutazione clinica come formulazione orale in associazione con uracile: l’uracile serve ad impedire la degradazione enzimatica del FU ed aumenta la biodisponibilità del farmaco dopo somministrazione orale.
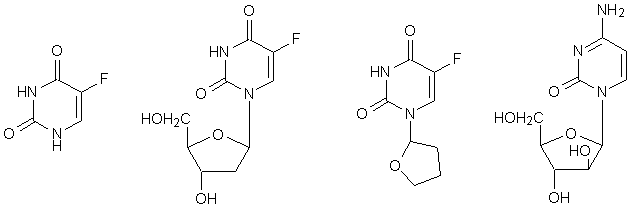
5-fluorouracile flossuridina fluorofuran Ara-C
Altro antimetabolita pirimidinico di grande interesse clinico è l’Ara-C (1-b-D-arabinofuranosilcitosina), un nucleoside ad attività antivirale formato dall’unione di una base fisiologica, la citosina, con uno zucchero non fisiologico, l’arabinosio. E’ anche detta citosin-arabinoside o citarabina.
Essa inibisce la conversione della citidinadifosfato in 2’-desossicitidinadifosfato ed ancora l’incorporazione della 2’-desossicitidinatrifosfato nel DNA, sostituendosi ad essa e fornendo un Ara-RNA spurio. Le vere forme attive dell’Ara-C sono le sue forme fosforilate e l’elevata attività antileucemica in vivo di questo farmaco è da mettere in relazione con l’elevata capacità mostrata dalle cellule leucemiche nel formare i nucleotidi corrispondenti all’Ara-C. Caratteristica peculiare dell’Ara-C rispetto agli altri antitumorali antileucemici è quella di essere attiva più contro le leucemie mielocitica e monocitica acuta e meno contro la leucemia linfocitica acuta.
1.3 Antagonisti purinici
Delle varie purine non naturali che sono state sintetizzate ed esaminate come antitumorali le più attive sono risultate la 6-mercaptopurina (MP) e la 6-tioguanina (TG).
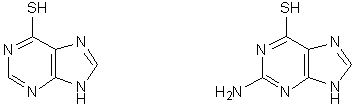
mercaptopurina (MP) tioguanina (TG)
Nel 50% dei casi di leucemia infantile acuta il trattamento con MP porta alla remissione completa della malattia, mentre la TG, come farmaco singolo, non presenta particolari vantaggi giacché ad una sua maggiore attività corrisponde una altrettanto maggiore tossicità.
La MP viene trasformata per via enzimatica nel corrispondente nucleoside monofosfato e, quindi, trifosfato. In tale forma viene incorporata negli acidi nucleici impedendo così la sintesi proteica. Inoltre i nucleosidi che da essa si formano inibiscono la sintesi della 5-fosforibosil-1-amina e la conversione della inosinamonofosfato (IMP) ad adenosinamonofosfato (AMP).
2. Farmaci provvisti di elevata reattività chimica ma privi di
specificità di azione (farmaci
alchilanti)
Al gruppo degli agenti alchilanti appartengono composti chimicamente reattivi che si combinano mediante legami covalenti con i centri nucleofili (gruppi aminici, tiolici, ossidrilici, fosforici…) presenti nei materiali biologici (proteine, acidi nucleici…), che vengono in tal modo danneggiati.
2.1 Mostarde azotate
La prima sostanza ad ottenere l’approvazione per il trattamento in sede clinica di talune forma di cancro è stata la clormetina (mecloretamina negli USA e mustina in UK). Con essa ha avuto inizio l’era della moderna chemioterapia antineoplastica. Sintetizzata nel 1935, è tuttora l’agente alchilante di elezione nella cura del morbo di Hodgkin, sia da sola che in associazione con altri chemioterapici antitumorali.
Dopo la mecloretamina alcune migliaia di mostarde azotate (caratterizzate dalla presenza del gruppo b-cloroetilaminico) sono state sintetizzate, ma solo poche di esse hanno superato il vaglio della sperimentazione clinica e sono usate attualmente in terapia. Tra queste ricordiamo il nitromin (forma latenziata della mecloretamina), il melfalan, la ciclofosfamide e suoi analoghi.
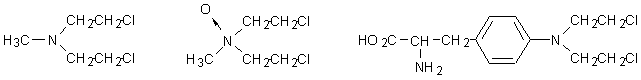
mecloretamina nitromin melfalan
ciclofosfamide trofosfamide ifosfamide
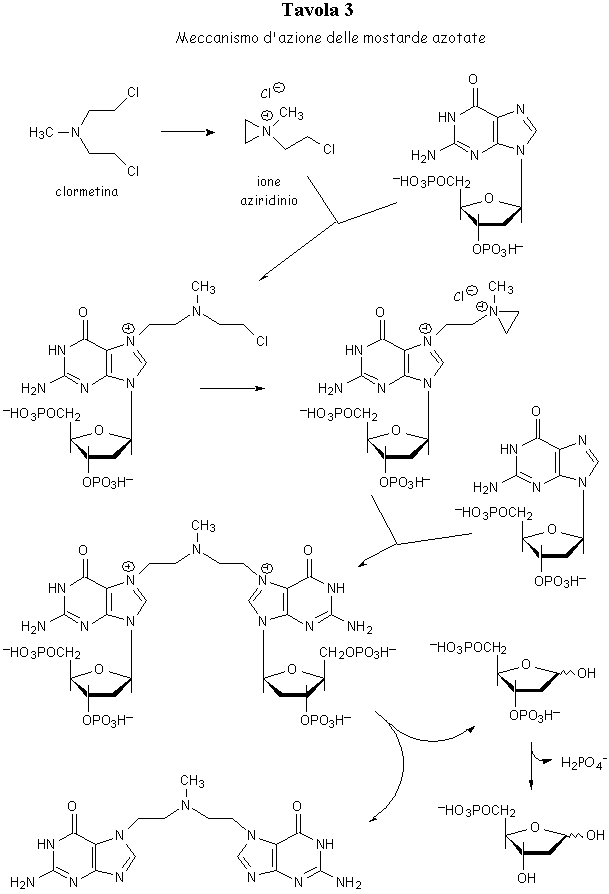
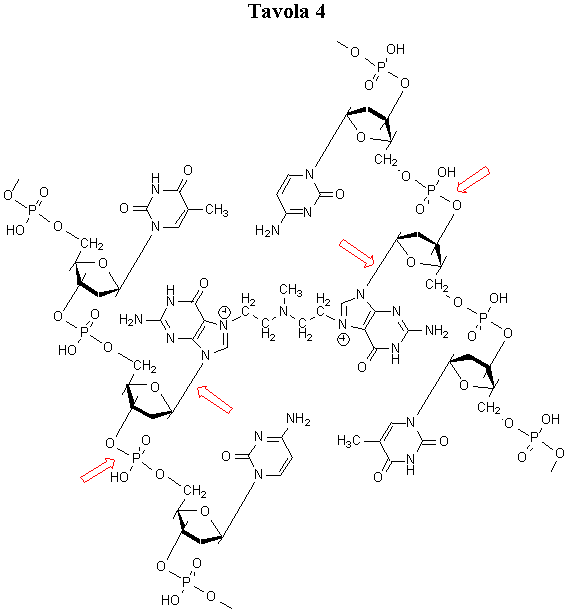
Per quanto attiene al meccanismo di azione delle mostarde azotate (Tavola 3) è stato provato che al pH fisiologico la mecloretamina cloridato viene prontamente trasformata in uno ione aziridinio, il vero agente alchilante altamente reattivo. Ambedue i gruppi cloroetilici reagiscono in tale maniera, realizzando per tappe una doppia alchilazione. Si ritiene che in vivo la dialchilazione avvenga prevalentemente a livello del DNA all’altezza della posizione N-7 della guanina, con formazione di un legame trasversale tra le due eliche complementari (Tavola 4). La formazione di un tale legame trasversale comporterebbe la successiva depurinazione e la rottura della catena polinucleotidica del DNA, che sarebbe così danneggiato e non più in grado di duplicarsi e di portare a termine la sintesi proteica.
Il nitromin è stato studiato per ovviare all’elevato potere vescicante che, per la sua elevata reattività chimica, la mecloretamina manifesta in sede di applicazione endovenosa. Il nitromin, inzialmente inattivo perché non può generare uno ione aziridinio, verrebbe trasformato per riduzione in vivo nella mostarda originaria, il vero agente alchilante.
Il melfalan fa parte delle mostarde azotate aromatiche, composti provvisti di minore reattività chimica e quindi meno tossici, al punto da poter essere usati per via orale. Sintetizzato con l’idea di ottenere un analogo di un aminoacido naturale (fenilalanina) che possedesse una maggiore selettività di azione nei confronti delle cellule neoplastiche, il melfalan trova applicazione per via orale come farmaco di elezione nel trattamento del mieloma multiplo. E’ anche utilizzato con eccellenti risultati nel trattamento post-chirurgico del cancro della mammella. Con la denominazione melfalan si indica l’enantiomero levogiro sintetizzato in Inghilterra, mentre l’altro isomero ottico od il corrispondente racemo, studiati in Russia, sono indicati col nome di sarcolisina.
Tutti i composti testè visti, con l’eccezione del nitromin, per la loro alta reattività chimica agiscono immediatamente una volta introdotti nell’organismo. Ciò significa che sia le cellule sane del corpo umano che quelle tumorali vengono indifferentemente attaccate dal gruppo alchilante e la eventuale selettività di azione del farmaco viene a dipendere esclusivamente dalla struttura portante (carrier). Queste considerazioni hanno indirizzato gli studiosi verso la messa a punto di composti che, una volta assunti, risultino inizialmente inattivi e vengano bioattivati solamente dopo aver raggiunto la sede di azione. In altre parole in essi il gruppo biologicamente attivo sarebbe presente all’inizio in forma latente inattiva, suscettibile però di bioattivazione selettiva a livello del tumore.
Un tale farmaco ideale non è stato ancora ottenuto; tuttavia la ciclofosfamide, il più usato in sede clinica dei chemioterapici alchilanti, costituisce un eccellente esempio di farmaco in forma latente. Dal punto di vista clinico la ciclofosfamide è la migliore mostarda di cui si disponga per curare carcinomi e sarcomi, leucemie e linfomi. E’ anche largamente impiegata nelle associazioni polichemioterapiche. Tra i molti analoghi e derivati della ciclofosfamide che sono stati successivamente studiati, due hanno raggiunto il mercato: la ifosfamide e la trofosfamide.
Anche la ciclofosfamide ed i suoi analoghi agiscono come la mecloretamina; essi però risultano meno tossici in quanto agirebbero solo dopo bioattivazione. La ciclofosfamide, inizialmente inattiva, verrebbe metabolizzata a livello del fegato con formazione di vari prodotti. Alcuni di essi, risultati inattivi, sono probabilmente prodotti di detossificazione; tra i rimanenti la vera forma biologicamente attiva non è stata tuttavia ancora individuata con certezza (Tavola 5).
2.2 Aziridine
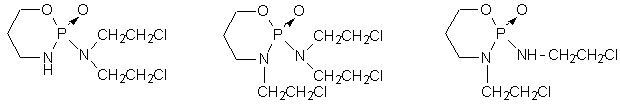
La constatazione che la vera forma biologicamente attiva delle mostarde azotate è il corrispondente ione aziridinio ha portato come conseguenza a studiare e sperimentare come agenti anticancro numerosissime aziridine. Il primo agente utile per via orale è stato il tiotepa, che resta il più importante dei farmaci aziridinici.
tiotepa mitomicina C
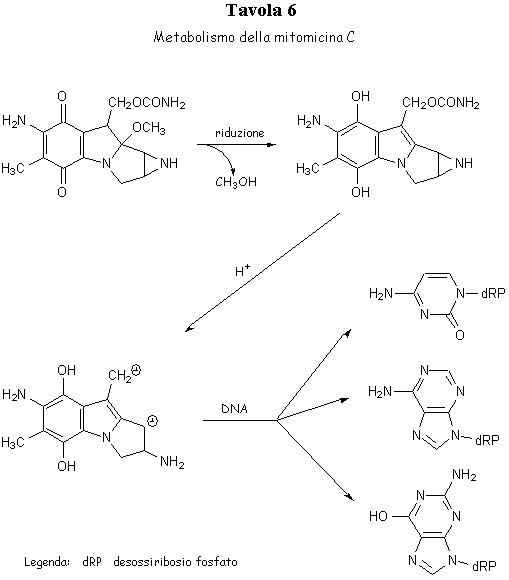
Un composto naturale molto interessante è la mitomicina C, antibiotico antitumorale scoperto nel 1962, nella cui struttura compaiono i raggruppamenti benzochinonico, aziridinico ed uretanico, la cui presenza in numerosi chemioterapici antitumorali sintetici era già in precedenza risultata determinante per il manifestarsi dell’attività biologica[PC1].
Nell’organismo infatti la mitomicina C viene ridotta nella forma diidrochinonica, che per successiva protonazione all’azoto aziridinico si trasforma in un agente bialchilante capace di legarsi, a livello del DNA, ai gruppi aminici dell’adenina e della citosina ed all’ossigeno della guanina (Tavola 6).
2.3 Altri alchilanti
Anche gli esteri metansolfonici e gli epossidi, analogamente alle mostarde azotate ed alle aziridine, possono agire da alchilanti. Esempio saliente di diestere metansolfonico è fornito dal busulfan. Questo agente anticancro è caratterizzato da una molecola bifunzionale simmetrica che ricorda nel meccanismo di azione la mecloretamina. Anch’esso infatti agisce formando legami trasversali tra le due eliche coassiali del DNA, presumibilmente a livello della guanina. Da notare che mentre gli omologhi inferiori del busulfan sono biologicamente inattivi, gli omologhi superiori sono ancora provvisti di attività di rilievo. Tuttavia il busulfan è l’unico estere metansolfonico entrato in terapia per il trattamento della leucemia mielogenica cronica.

busulfan pipobroman
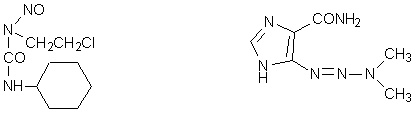
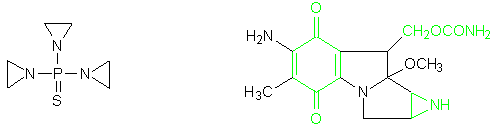
lomustina (CCNU) dacarbazina (DTIC)
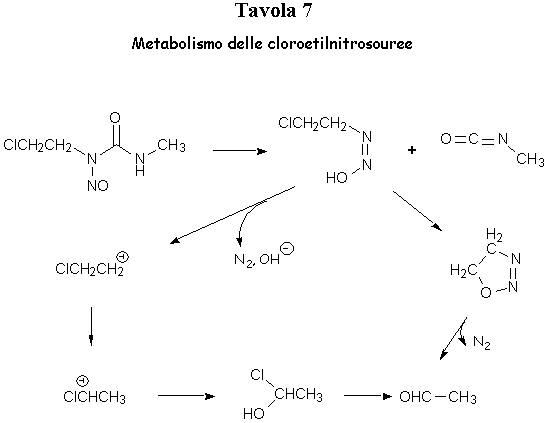
Tutti i farmaci sin qui trattati contengono due gruppi alchilanti ed in effetti questa è la condizione ideale per avere la massima attività biologica, tuttavia anche agenti alchilanti monofunzionali risultano attivi come, ad esempio, le N-cloroetilnitrosouree, quali la lomustina (CCNU), attiva per via orale su vari tumori solidi ed usata in particolare nel trattamento di taluni tumori del cervello e, soprattutto in combinazione con FU, nei tumori del colon.
Nell’organismo le cloroetilnitrosouree vengono degradate metabolicamente con formazione di aldeide acetica, cloroetanolo, uree sostituite, carbocationi ed isocianati (Tavola 7). Questi due ultimi sono i gruppi responsabili dell’attività antitumorale e della tossicità manifestate dalle nitrosouree, farmaci capaci di alchilare gli acidi nucleici e di carbamoilare le proteine dell’organismo. Sebbene formalmente presentino una sola funzione alchilante, le nitrosouree in realtà si comportano da bialchilanti in virtù della formazione del carbocatione ClCH2CH2(+).
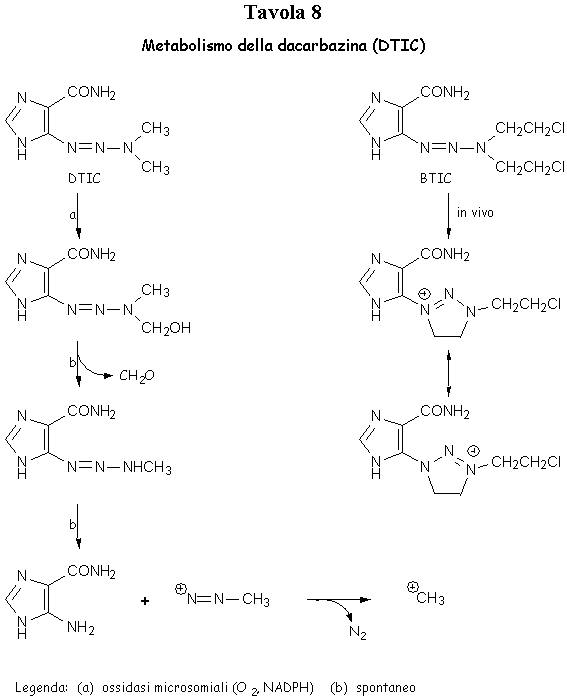
Anche la dacarbazina o DTIC, un derivato triazenoimidazolico, si comporta da agente alchilante. Questo composto, che trova utilizzazione come farmaco di elezione nel trattamento del melanoma melanotico e mostra sinergismo di azione con adriamicina nel trattamento dei sarcomi dei tessuti molli, nell’organismo subisce una serie di trasformazioni da parte delle ossidasi microsomiali epatiche con formazione del carbocatione metilico proveniente dalla decomposizione dello ione metildiazonio instabile (Tavola 8).
3. Farmaci che complessano il DNA
Questo gruppo di farmaci comprende composti, tutti appartenenti alla classe degli antibiotici, capaci per la loro particolare struttura chimica di intercalarsi periodicamente tra due basi adiacenti del DNA, stabilendo con esse una unione, mediante legami idrogeno, per lo più nel giro minore della struttura bielicoidale del DNA. Come conseguenza di questa intercalazione di un corpo estraneo elettrostaticamente attivo nel contesto del DNA, viene ostacolato il lavoro della RNA-polimerasi DNA-dipendente con impedimento della trascrizione.
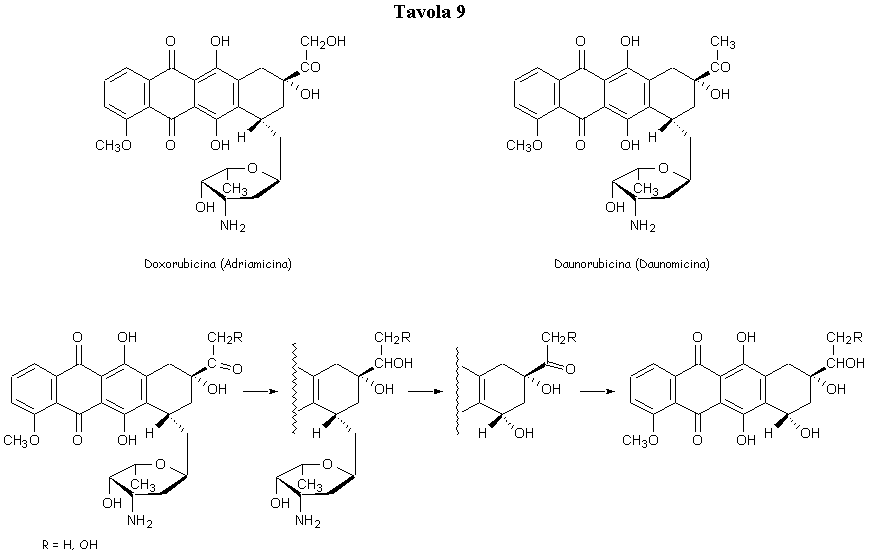
Dei vari antibiotici di questa classe, le antracicline adriamicina e daunomicina sono i più importanti sul piano clinico, con l’adriamicina, più attiva, particolarmente indicata nella cura dei tumori solidi, dei linfomi e delle leucemie acute.
Ambedue gli antibiotici vengono metabolizzati in vivo con riduzione della funzione chetonica del gruppo acetile. Gli alcoli corrispondenti risultano attivi al pari dei chetoni di partenza e contribuiscono anch’essi all’attività biologica. Successivamente si mette in libertà, per idrolisi enzimatica a livello del legame glucosidico, la porzione agliconica inattiva (Tavola 9). Entrambi questi antibiotici sono caratterizzati da cardiotossicità (arresto cardiaco).
4. Inibitori della mitosi
Un altro gruppo molto importante di farmaci antitumorali è quello costituito dagli inibitori della mitosi cellulare.
Hanno manifestato attività antimitotica gli alcaloidi della Vinca, la colchicina e suoi derivati, la podofillotossina ed analoghi, la griseofulvina, il paclitaxel e congeneri. Tutti questi composti sono capaci di arrestare il processo mitotico allo stadio della metafase con alterazione del fuso acromatico.
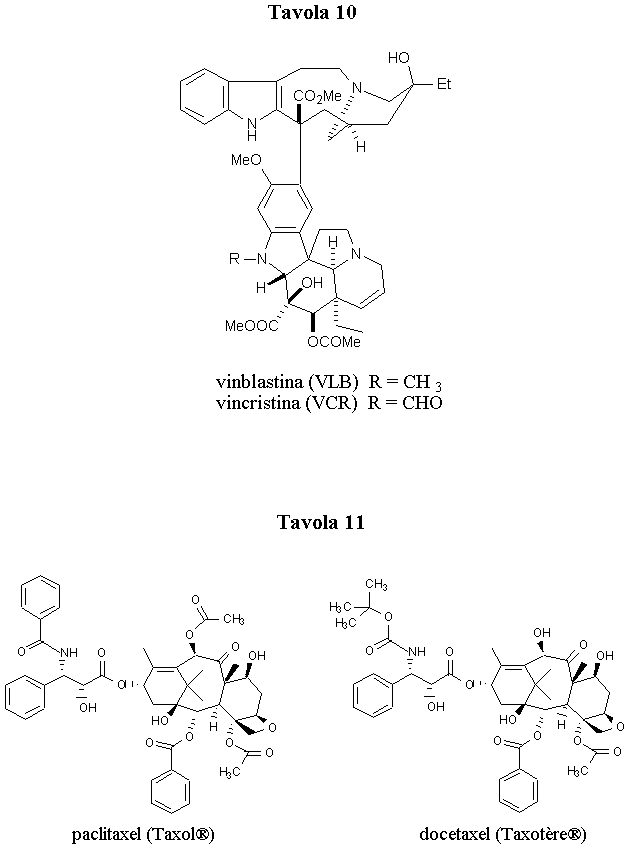
In base all’esatto meccanismo di azione, essi vengono distinti in tre gruppi principali: composti che stabilizzano i microtubuli, composti che si legano al sito di legame degli alcaloidi della Vinca e composti che agiscono sul sito di binding della colchicina. I composti del primo gruppo inibiscono la depolimerizzazione dei microtubuli a tubulina legandosi ad essi e stabilizzandoli, mentre i composti degli altri due gruppi inibiscono la polimerizzazione della tubulina a microtubuli. Nonostante queste differenze, la perturbazione dell’equilibrio dinamico tubulina-microtubuli conduce allo stesso risultato finale: arresto della divisione cellulare a livello di metafase e induzione di apoptosi. Il paclitaxel ed il docetaxel, introdotti di recente in terapia, sono i capostipiti della famiglia dei taxani antimitotici, ma negli ultimi anni sono stati scoperti altri prodotti naturali capaci di stabilizzare i microtubuli, come gli epotiloni e l’eleuterobina (ed il corrispondente aglicone sarcodictina A). Tutti questi composti interagiscono con i microtubuli legandosi allo stesso sito di legame dei taxani.
Attualmente solo gli alcaloidi della Vinca (vinblastina e vincristina) (Tavola 10) ed il paclitaxel (Taxol®), col suo analogo più idrosolubile docetaxel (Taxotere®) (Tavola 11), sono impiegati in terapia. Gli alcaloidi della Vinca sono i farmaci di elezione in tutti i tipi di associazioni chemioterapiche antitumorali. I due taxani di uso terapeutico rappresentano la più recente acquisizione nel settore degli antitumorali e mostrano attività impressionante contro forme avanzate di tumore delle ovaie, della mammella, del polmone, dell’esofago, della vescica.
5. Ormoni
I primi tipi di cancro ad essere trattati mediante somministrazione di ormoni sono stati il cancro della mammella e quello della prostata.
Il concetto guida in questo tipo di terapia è che le cellule neoplastiche di un organo sensibile agli ormoni possono, almeno in qualche stadio dell’evoluzione neoplastica, essere sottoposte ad un controllo ormonale. In effetti una alterazione dell’ambiente ormonale produce per lo più una remissione dei tumori nei casi di cancro della mammella, della prostata e dell’endometrio.
Il cancro della mammella è una delle forme più diffuse di cancro umano e causa ogni anno la morte di molte donne. La resezione chirurgica o mastectomia è il primo intervento in caso di diagnosi di tumore maligno alla mammella; ad esso fa seguito la cobaltoterapia come terapia coadiuvante al fine di distruggere eventuali cellule neoplastiche ancora presenti ed evitare le metastasi.
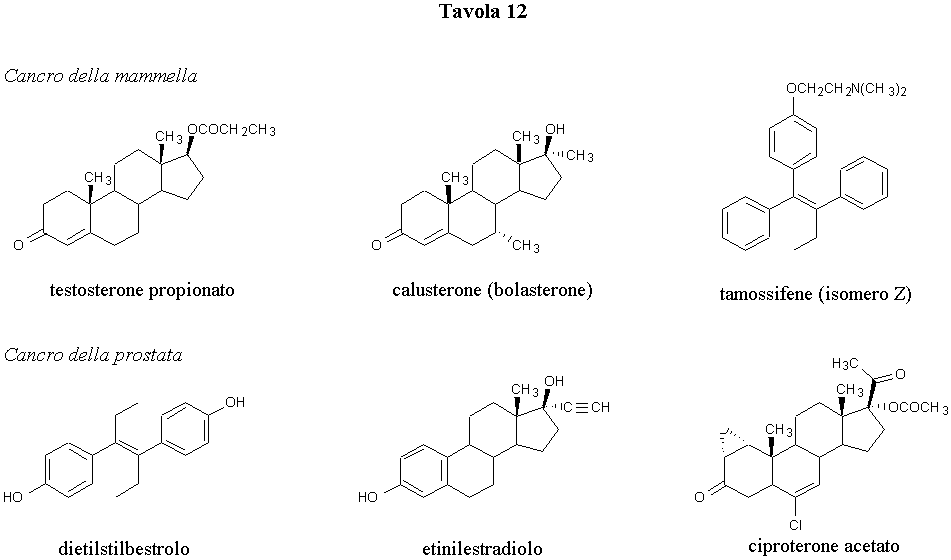
Dopo l’isolamento del testosterone e la successiva constatazione che esso manifestava azione antagonista nei confronti degli effetti fisiologici degli estrogeni, si tentò una terapia del cancro mammario sulla base dell’assunzione che questa forma di cancro fosse provocata, od almeno favorita, da agenti estrogeni. Si è così introdotta nell’uso clinico la terapia androgena per curare il cancro della mammella e ad essa ha fatto seguito successivamente il trattamento con estrogeni (dietilstilbestrolo, etinilestradiolo) per combattere lo stesso tipo di tumore nella donna in periodo di post-menopausa (Tavola 12).
Il farmaco classico ad attività androgena usato in sede clinica per il trattamento del cancro mammario è il testosterone propionato, che però possiede marcati effetti virilizzanti ovviamente indesiderati da parte delle donne. Pertanto la ricerca si è orientata verso lo studio di derivati del testosterone ad attività prevalentemente anabolizzante. Un recente acquisto terapeutico è il calusterone, uno dei più attivi ormonoidi anticancro, usato per via orale e risultato provvisto di scarsa attività androgena indesiderata. Il calusterone è il più usato degli steroidi nella terapia ormonica primaria e secondaria del cancro della mammella.
Alte dose di estrogeni in genere sono risultate più attive degli androgeni nella terapia ormonale del cancro della mammella nelle donne in periodo di post-menopausa. A questo scopo vengono utilizzati indifferentemente il dietilstilbestrolo e l’etinilestradiolo. E’ da rimarcare che solo alte dosi di estrogeni inibiscono il tumore mammario, mentre piccole dosi di tali ormoni addirittura favoriscono la sua crescita.
Un approccio alternativo si basa sull’impiego di antiestrogeni. Il tamossifene viene usato in più di 70 Paesi per il trattamento dei tumori in fase avanzata (metastatica). Esso provoca un blocco G1 nel ciclo cellulare delle cellule neoplastiche, che viene a sua volta ripristinato dall’estradiolo (ormone estrogeno): perciò il tamossifene, che deve essere considerato più un farmaco tumoristatico che tumoricida, è raccomandato nel trattamento del cancro al seno delle donne in menopausa. Da notare che dei due possibili isomeri geometrici del tamossifene, solo la forma Z (cis) si comporta da antiestrogeno, mentre l’isomero E (trans) funziona da estrogeno e non viene impiegato nella cura del cancro mammario.
Gli estrogeni sono anche usati nella terapia del cancro della prostata, una forma maligna molto diffusa tra gli uomini. La crescita ed il funzionamento della prostata è regolato dall’azione degli ormoni androgeni, di conseguenza il fine della terapia anticancro ormonica in questo caso è quello di antagonizzare gli stimoli prodotti dagli ormoni androgeni.
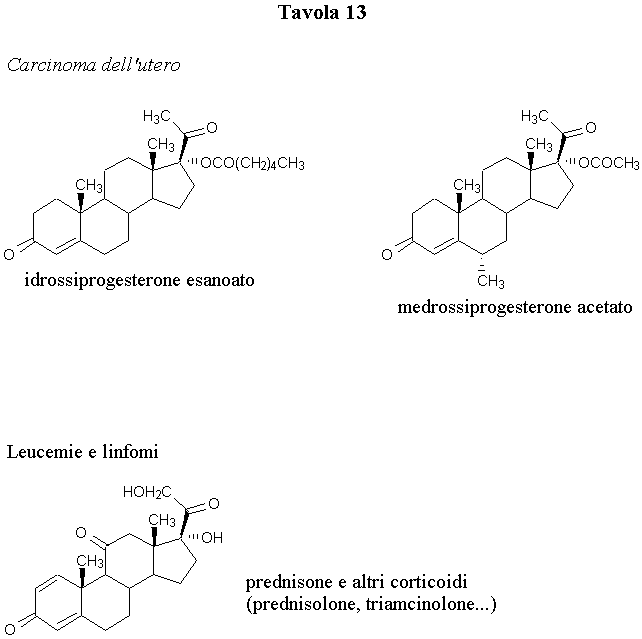
I farmaci progestinici, cioè i farmaci riferibili per struttura chimica ed attività biologica al progesterone, hanno fornito risultati notevoli nella terapia del carcinoma primario dell’utero (endometrio). Il 30-35% delle pazienti in genere risponde favorevolmente alla cura con 17a-idrossiprogesterone esanoato o con medrossiprogesterone acetato (Tavola 13).
Il ciproterone è anch’esso un derivato progestinico, ma la sua maggiore utilizzazione riguarda il cancro della prostata.
I corticosteroidi, anch’essi riconducibili chimicamente al progesterone, trovano impiego principalmente nelle forme leucemiche e nei linfomi. I corticosteroidi naturali, come il cortisone e l’idrocortisone, hanno ormai ceduto il passo ai glucocorticoidi semisintetici (prednisone, prednisolone, desametazone, triamcinolone).
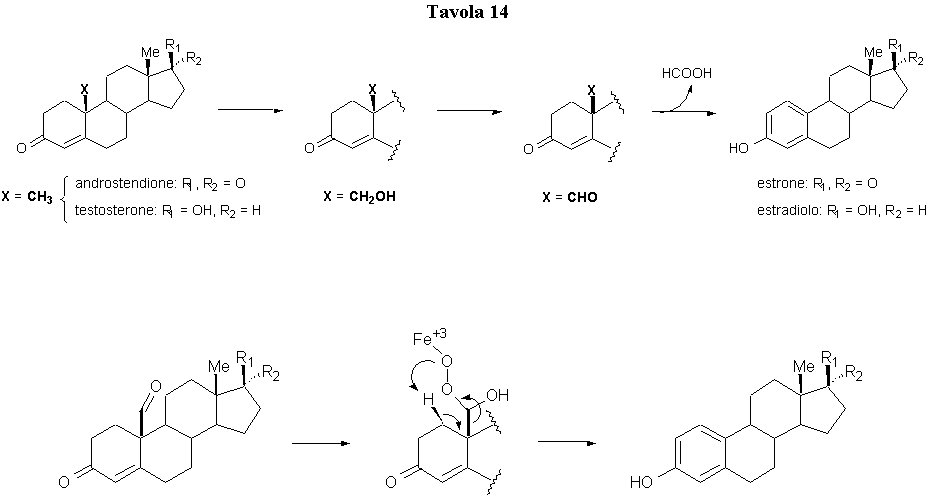
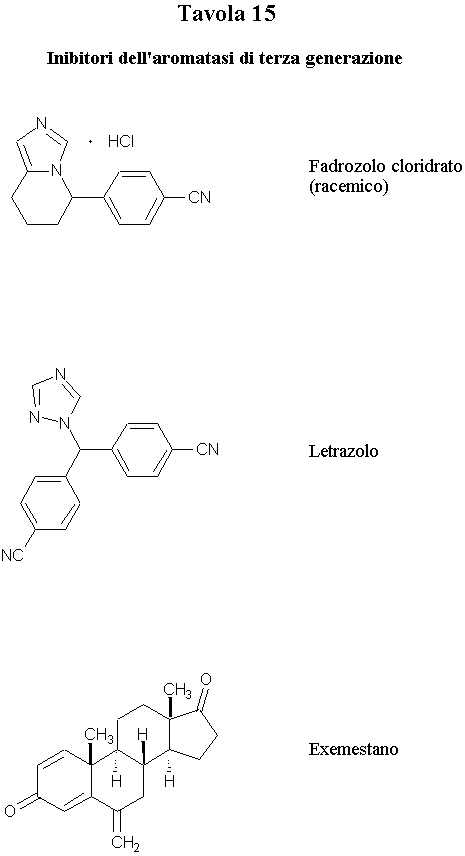
Recenti scoperte nella terapia endocrina del tumore della mammella hanno dimostrato i benefici effetti dell’inibizione dell’enzima aromatasi, un enzima citocromo P450-dipendente che catalizza lo stadio finale della biosintesi degli estrogeni (Tavola 14). L’inibizione di tale enzima può essere ottenuta mediante analoghi steroidici del substrato naturale dell’enzima oppure con inibitori non steroidei che sono strutturalmente correlati con gli antifungini azolici (Tavola 15). Fadrozolo, letrazolo ed exemestano sono tutti inibitori dell’aromatasi di terza generazione attivi per somministrazione orale.
Il fadrozolo cloridrato (racemico) è stato introdotto in terapia nel 1995 per il trattamento del cancro della mammella post-menopausale. E’ un inibitore potente e specifico dell’aromatasi privo di effetti di tipo androgeno ed estrogeno e di altri significativi effetti collaterali. Esercita la sua azione coordinando il ferro del nucleo porfirinico del citocromo P450 probabilmente attraverso la porzione imidazolica. Questa forte interazione impedisce che il ferro possa legare l’ossigeno molecolare ed inattiva reversibilmente l’enzima.
Il letrazolo (1996) è il più selettivo tra gli inibitori dell’aromatasi studiati finora. Risulta circa 10.000 volte più potente dell’aminoglutetimide, che è stato il primo inibitore dell’aromatasi ad essere scoperto. Presenta una potente azione antitumorale ed è privo di effetti collaterali indesiderati.
L’exemestano è stato sviluppato in Italia dalla Farmitalia Carlo Erba ed è stato introdotto per la prima volta da Pharmacia nel 2000 per il trattamento di tumori estrogeno-dipendenti e del cancro della mammella post-menopausale. E’ un nuovo composto steroideo di sintesi strutturalmente riconducibile al substrato naturale per la biosintesi degli estrogeni, l’androstandione. L’exemestano è un inibitore irreversibile dell’aromatasi e presenta attività endocrina minima. Si tratta del primo rappresentante steroideo degli inibitori dell’aromatasi di terza generazione.
6. Enzimi
La L-aparaginasi costituisce il primo esempio di farmaco antitumorale il cui meccanismo di azione non comporti l’inibizione dell’accrescimento e della moltiplicazione cellulare. Questo enzima catalizza la scissione della L-asparagina in acido aspartico ed ammoniaca e priva quindi le cellule asparagino-dipendenti di un aminoacido indispensabile alla loro vita.
Come è noto, per le cellule sane questo non è un aminoacido essenziale; al contrario, la maggior parte delle cellule leucemiche ed un numero molto limitato di tumori solidi avrebbero perduto la facoltà di sintetizzare l’asparagina, la cui carenza provocherebbe perciò l’estinzione di diversi e importanti processi metabolici con conseguente distruzione delle cellule tumorali stesse.
La L-asparaginasi, prodotta su larga scala in Germania e negli USA usando come fonte l’Escherichia coli, presenta per ovvie ragioni uno spettro di azione limitato e trova applicazione quasi esclusiva nel trattamento delle leucemie acute linfoblastica e mieloblastica.
7. Farmaci vari
Molti altri farmaci, alcuni dei quali classificabili ancora nelle precedenti classi, sono reperibili in commercio all’estero ed altri, sotto indagine, costituiscono una speranza per il futuro della chemioterapia antineoplastica (Tavola 16).
![]()
cis-platino
Uno dei più interessanti tra quelli usati negli ultimi anni è senza dubbio il cis-platino o cis-diclorodiaminoplatino. Questo composto agisce a livello del DNA alterandone la struttura ed inibendo i sistemi di riparazione del DNA presenti nell’organismo. Esso, come del resto tutti i citostatici che attaccano il DNA, è provvisto di proprietà mutagene. Da solo produce la regressione totale o parziale di vari tipi di cancro: testicolare, polmonare, dello stomaco, delle ovaie, dell’utero e della cervice. Agisce anche su carcinomi epidermoidi e melanomi. Viene usato soprattutto in associazione con altri oncostatici (ciclofosfamide, adriamicina, bleomicina, vincristina), scelti opportunamente a seconda del tipo di cancro da curare.
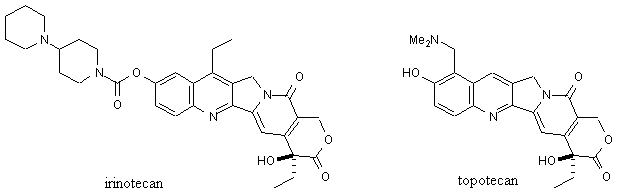
Due derivati semisintetici ed idrosolubili della camptotecina, l’irinotecan e il topotecan, sono stati introdotti in terapia nel 1994 e nel 1996, rispettivamente, quali nuovi farmaci antineoplastici inibitori della topoisomerasi-I, un enzima cellulare deputato al mantenimento della struttura del DNA durante i processi di traslazione, trascrizione e mitosi. Nonostante l’elevata analogia strutturale, i due composti presentano uno spettro di attività antitumorale ed effetti collaterali decisamente diversi. L’irinotecan (un profarmaco che in vivo subisce l’idrolisi del gruppo carbammato per dare il metabolita attivo, 1000 volte più potente del precursore) è usato per il trattamento del cancro colorettale. Sebbene meno tossico della camptotecina, provoca gravi forme di diarrea, nausea/vomito, leucopenia, alopecia. Il topotecan non è un profarmaco ed è utilizzato nel carcinoma ovarico e polmonare. Provoca neutropenia.